


Mapping for Segmented Viruses Pipeline
Introduction
The pipeline "Mapping for Segmented Viruses" performs mapping of genome fragments from segmented viruses using multiple references, which allows mapping of each segment to the most appropriate reference. This pipeline outputs both alignment files and the whole multifasta file.

Run Mapping for Segmented Viruses Pipeline
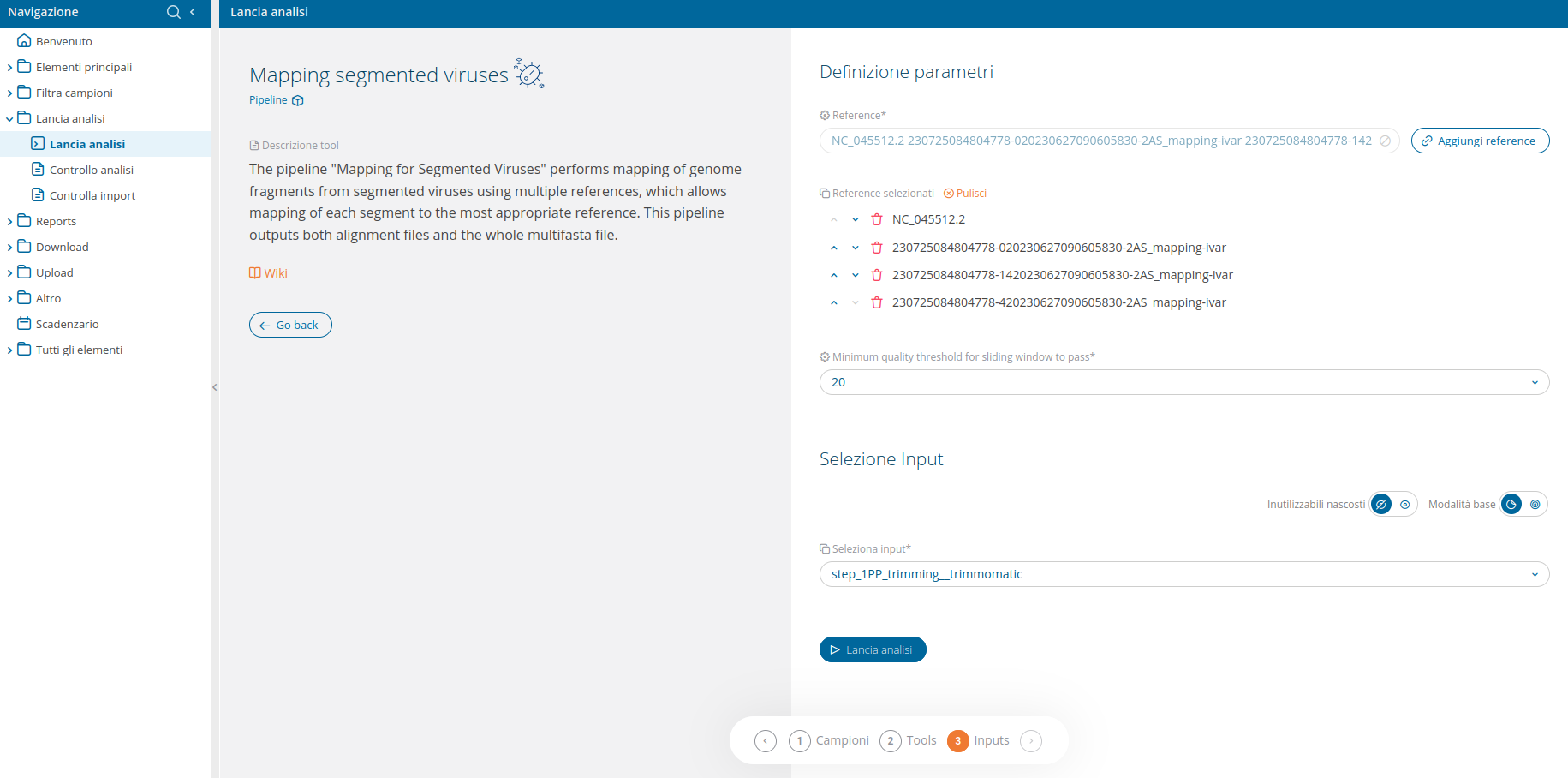
The filter at the top of the run analyses page allows to display only the pipelines. Once the Mapping for Segmented Viruses pipeline has been selected, a confirmation interface will be displayed.
The Mapping for Segmented Viruses pipeline performs mapping with Ivar (2AS_mapping__ivar) for all genome fragments from segmented viruses provided by the user.
Accepted inputs can be from pre-processing analyses:
- depleted reads (step_1PP_hostdepl)
- downsampled reads (step_1PP_downsampling)
- trimmed reads (step_1PP_trimming)
- filtered reads (step_1PP_filtering)
The input selection UI delivers an advanced input selection mode, to allow selection of all types of supported input files at once.
A link to Check analysis will be created after launching the requested pipeline. The system will notify the user after a succesful pipeline launch and once execution has ended.
Multiple References
All required reference files will have to be provided by the user, through the dedicated reference selection wizard (please refer to the corresponding Wiki section on the run analysis topic).
Results
Output files from the "Mapping for Segmented Viruses" pipeline are the same as those produced by the single analyses included in it, in the same directory structure. Please refer to the corresponding 2AS_mapping Wiki section. On top of those files, the pipelina also outputs a multifasta file (more details in the table below).
| File | Description | Location |
|---|---|---|
| multifasta file containing all the fragment sequences mapped to the appropriate references | ||
| warnings.log | warning text file with information about incorrect references | main directory |
