


2AS_mapping
Introduction
2AS_mapping performs the assembly with reference (i.e. mapping) of nucleotide sequences.

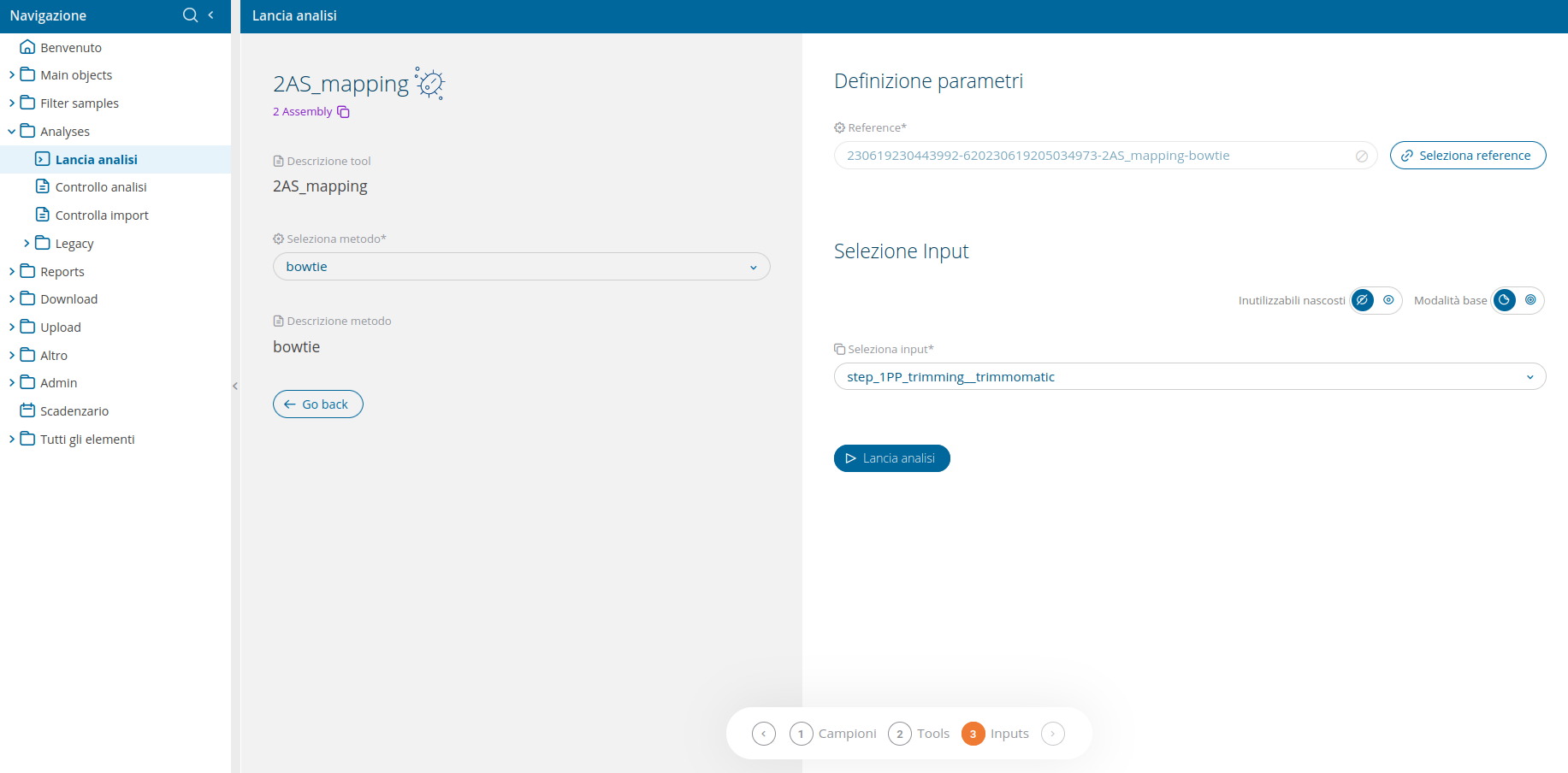
Run Analysis 2AS_mapping
Once the analysis 2AS_mapping has been selected from the run analyses interface, the user will be able to select which bioinformatic tool to use. The available tools are:
- bowtie2 - Fast and sensitive read alignment
- ivar - computational package for viral amplicon-based sequencing
Note: In case the software "snippy" is chosen, samples will need to have an associated 4AN_genes output files from "Prokka".
To select the mandatory reference genome, use the "Select reference" button in the input selection wizard. This will open a pop-up table, listing 2 different kind of sequences, both usable as reference:
- reference fasta files;
- consensus sequence or de novo assembly of a sample available in Cohesive.
With the tool "Ivar" it's possible to select multiple references (please consult the "Multiple references" section).
The input selection UI delivers an advanced input selection mode, to allow selection of all types of supported input files at once.
Accepted inputs can be from:
A link to Check analysis will be created after launching the requested analysis. The system will notify the user after a succesful analysis launch and once execution has ended.
Multiple references
If 2AS_mapping is performed with Ivar, it will be possible to select more than one reference, as shown in the image below. A guide to the multiple reference selection system is available at the corresponding section of the run analysis Wiki.
Output directory
Please refer to Cohesive's specific Wiki page for information on file download.
The output directory is available at the link in the download page or at the link presente in the analysis' summary card, and will have the following structure: results > YEAR > ID > 2AS_mapping > DSXXXXXXXX-DTXXXXXX_bowtie2. The last directory's suffix will be replaced with the name of the chosen tool. At that path there will be 2 directories:
- meta: ("metadata") contains log and configuration files.
- result: contains the analysis' output files.
The tables below list files produced by 2AS_mapping's available tools.
Bowtie2
| File | Description | Location |
|---|---|---|
| DSXXXXXXXX-DTXXXXXX_ID_bowtie_REFID.fasta | consensus file | result directory |
| DSXXXXXXXX-DTXXXXXX_ID_bowtie_REFID.sorted.bam | bam (Binary Alignment Map) format alignment file | result directory |
| DSXXXXXXXX-DTXXXXXX_ID_bowtie_REFID.sorted.bam.bai | bai (bam file's index) file | result directory |
| DSXXXXXXXX-DTXXXXXX_ID_bowtie_REFID.var.flt.vcf | vcf (variant calling format) file with identified varaints | result directory |
| DSXXXXXXXX-DTXXXXXX_ID_bowtie_REFID_coverage_plot.png | coverage distribution graph | result directory |
Ivar
Note: Ivar's execution consists of "Snippy", "Samtools" and "Ivar" tools execution.
| File | Description | Location |
|---|---|---|
| DSXXXXXXXX-DTXXXXXX_ID_ivar_REFID.fasta | consensus sequence from Ivar | result directory |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.aligned.fa | reference with - in positions with sequencing depth = 0and depth's N between 0 and the minimum number of reads considered for site coverage (no variants in this file) |
result directory |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.bam | bam (Binary Alignment Map) format alignment file | result directory |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.bam.bai | bai (bam file's index) file | result directory |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.bed | bed (Browser Extensible Data) file | result directory |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.consensus.fa | reference genome with representation of all variants | result directory |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.consensus.subs.fa | reference genome with representation of substitution variants | result directory |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.txt | snippy run summary | result directory |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.tab | variant table in tsv format | result directory |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.csv | variant table in csv format | result directory |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.filt.vcf | variants filtered by Freebayes | result directory |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.raw.vcf | variants not filtered by Freebayes | result directory |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.subs.vcf | table of substitution variants in vcf format | result directory |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.gff | variants in GFF3 format | result directory |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.html | html version of the .tab table of variants | result directory |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.vcf | Snippy's output file with identified variants in vcf format | result directory |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.vcf.gz | snippy's vcf output file (archive) | result directory |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID.vcf.gz.csi | bcftools index of vcf.gz file | result directory |
| DSXXXXXXXX-DTXXXXXX_ID_vdsnippy_REFID_coverage_plot.png | coverage distribution graph | result directory |
For further information about Snippy's output files, file formats and contents, please refer to Snippy's official manual.
Data visualization
Alignment of reads on the reference genome can be visualized with specific softwares (i.e. Tablet, BioEdit e uGene), which are able to read bam e bam.bai files:
- Tablet (GNU/Linux, macOS, Windows): https://ics.hutton.ac.uk/tablet/download-tablet/;
- BioEdit (Windows): https://thalljiscience.github.io/;
- uGene (GNU/Linux, macOS, Windows): http://ugene.net/ugene/.
